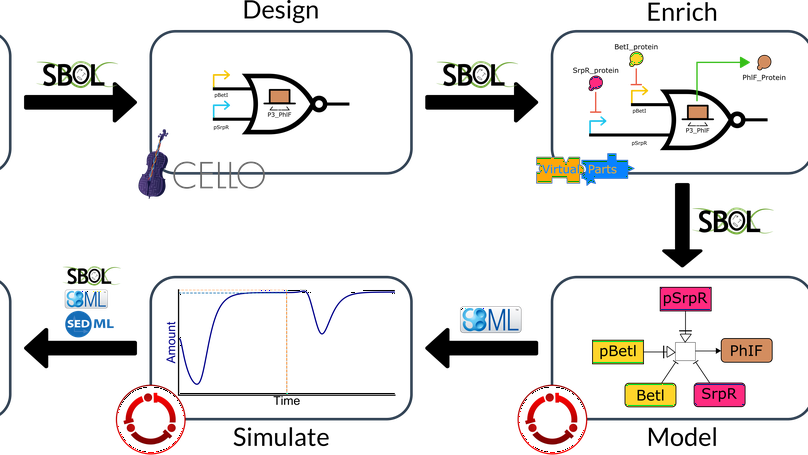
Synthetic biology builds upon genetics, molecular biology, and metabolic engineering by applying engineering principles to the design of biological systems. When designing a synthetic system, synthetic biologists need to exchange information about multiple types of molecules, the intended behavior of the system, and actual experimental measurements. The Synthetic Biology Open Language (SBOL) has been developed as a standard to support the specification and exchange of biological design information in synthetic biology, following an open community process involving both wet bench scientists and dry scientific modelers and software developers, across academia, industry, and other institutions. This document describes SBOL 3.0.0, which condenses and simplifies previous versions of SBOL based on experiences in deployment across a variety of scientific and industrial settings. In particular, SBOL 3.0.0, (1) separates sequence features from part/sub-part relationships, (2) renames Component Definition/Component to Component/Sub-Component, (3) merges Component and Module classes, (4) ensures consistency between data model and ontology terms, (5) extends the means to define and reference Sub-Components, (6) refines requirements on object URIs, (7) enables graph-based serialization, (8) moves Systems Biology Ontology (SBO) for Component types, (9) makes all sequence associations explicit, (10) makes interfaces explicit, (11) generalizes Sequence Constraints into a general structural Constraint class, and (12) expands the set of allowed constraints.